Early stopping is a technique that is very often used when training neural networks, as well as with some other iterative machine learning algorithms. The idea is quite intuitive - let us measure the performance of our model on a separate validation dataset during the training iterations. We may then observe that, despite constant score improvements on the training data, the model's performance on the validation dataset would only improve during the first stage of training, reach an optimum at some point and then turn to getting worse with further iterations.
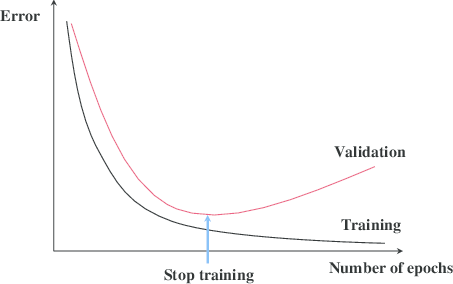
The early stopping principle
It thus seems reasonable to stop training at the point when the minimal validation error is achieved. Training the model any further only leads to overfitting. Right? The reasoning sounds solid and, indeed, early stopping is often claimed to improve generalization in practice. Most people seem to take the benefit of the technique for granted. In this post I would like to introduce some skepticism into this view or at least illustrate that things are not necessarily as obvious as they may seem from the diagram with the two lines above.
How does Early Stopping Work?
To get a better feeling of what early stopping actually does, let us examine its application to a very simple "machine learning model" - the estimation of the mean. Namely, suppose we are given a sample of 50 points  from a normal distribution with unit covariance and we need to estimate the mean
from a normal distribution with unit covariance and we need to estimate the mean  of this distribution.
of this distribution.
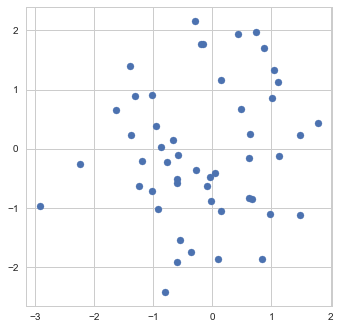
Sample
The maximum likelihood estimate of can be found as the point which has the smallest sum of squared distances to all the points in the sample. In other words, "model fitting" boils down to finding the minimum of the following objective function:
![\[f_\mathrm{train}(\mathrm{w}) := \sum_{i=1}^{50} \Vert \mathbf{x}_i - \mathbf{w}\Vert^2\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-4a4ff04b2699f9033f5d09897e4dfc5e_l3.png "Rendered by QuickLaTeX.com")
As our estimate is based on a finite sample, it, of course, won't necessarily be exactly equal to the true mean of the distribution, which I chose in this particular example to be exactly (0,0):
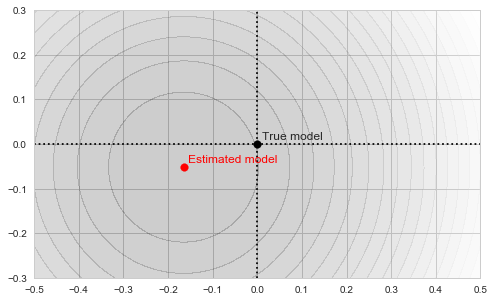
Sample mean as a minimum of the objective function
The circles in the illustration above are the contours of the objective function, which, as you might guess, is a paraboloid bowl. The red dot marks its bottom and is thus the solution to our optimization problem, i.e. the estimate of the mean we are looking for. We may find this solution in various ways. For example, a natural closed-form analytical solution is simply the mean of the training set. For our purposes, however, we will be using the gradient descent iterative optimization algorithm. It is also quite straightforward: start with any point (we'll pick (-0.5, 0) for concreteness' sake) and descend in small steps downwards until we reach the bottom of the bowl:
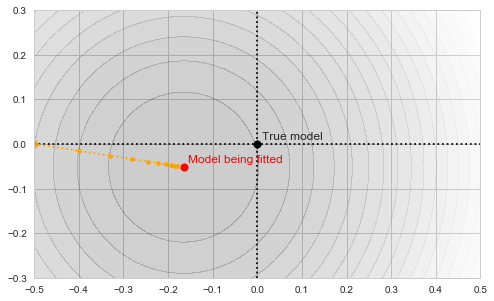
Gradient descent
Let us now introduce early stopping into the fitting process. We will split our 50 points randomly into two separate sets: 40 points will be used to fit the model and 10 will form the early stopping validation set. Thus, technically, we now have two different objective functions to deal with:
![\[f_\mathrm{fit}(\mathrm{w}) := \sum_{i=1}^{40} \Vert \mathbf{x}_i - \mathbf{w}\Vert^2\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-5adb94ca761b724a00c38b830a6bd831_l3.png "Rendered by QuickLaTeX.com")
and
![\[f_\mathrm{stop}(\mathrm{w}) := \sum_{i=41}^{50} \Vert \mathbf{x}_i - \mathbf{w}\Vert^2.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-8ac10bff72d2e3925e207369119b4537_l3.png "Rendered by QuickLaTeX.com")
Each of those defines its own "paraboloid bowl", both slightly different from the original one (because those are different subsets of data):
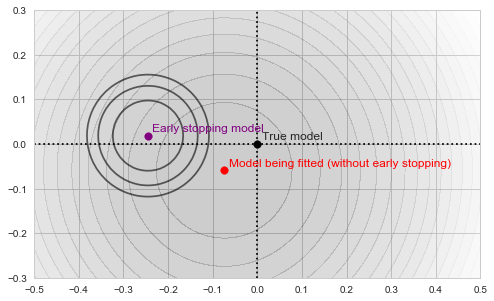
Fitting and early stopping objectives
As our algorithm descends towards the red point, we will be tracking the value of  at each step along the way:
at each step along the way:
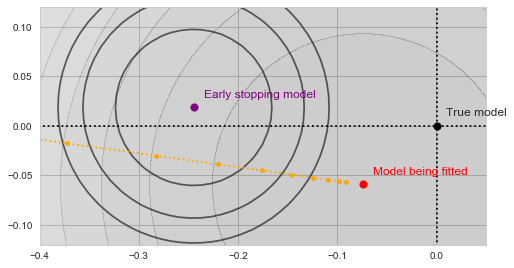
Gradient descent with validation
With a bit of imagination you should see on the image above, how the validation error decreases as the yellow trajectory approaches the purple dot and then starts to increase after some point midway. The spot where the validation error achieves the minimum (and thus the result of the early stopping algorithm) is shown by the green dot on the figure below:
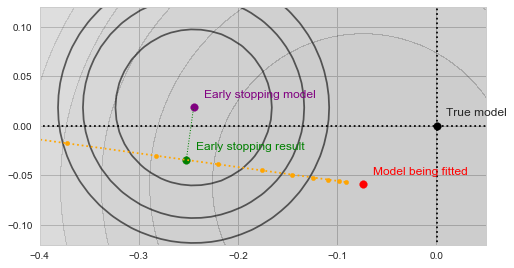
Early stopping
In a sense, the validation function now acts as a kind of a "guardian", preventing the optimization from converging towards the bottom of our main objective. The algorithm is forced to settle on a model, which is neither an optimum of  nor of . Moreover, both and use less data than
nor of . Moreover, both and use less data than  , and are thus inherently a worse representation of the problem altogether.
, and are thus inherently a worse representation of the problem altogether.
So, by applying early stopping we effectively reduced our training set size, used an even less reliable dataset to abort training, and settled on a solution which is not an optimum of anything at all. Sounds rather stupid, doesn't it?
Indeed, observe the distribution of the estimates found with (blue) and without (red) early stopping in repeated experiments (each time with a new random dataset):
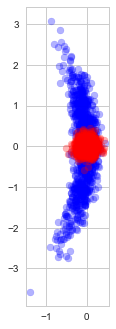
Solutions found with and without early stopping
As we see, early stopping greatly increases the variance of the estimate and adds a small bias towards our optimization starting point.
Finally, let us see how the quality of the fit depends on the size of the validation set:
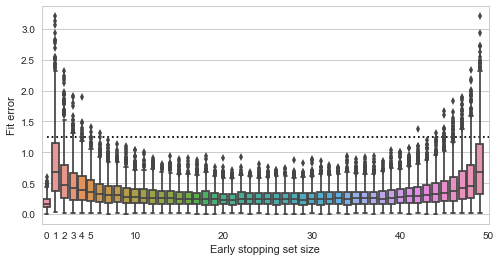
Fit quality vs validation set size
Here the y axis shows the squared distance of the estimated point to the true value (0,0), smaller is better (the dashed line is the expected distance of a randomly picked point from the data). The x axis shows all possible sizes of the validation set. We see that using no early stopping at all (x=0) results in the best expected fit. If we do decide to use early stopping, then for best results we should split the data approximately equally into training and validation sets. Interestingly, there do not seem to be much difference in whether we pick 30%, 50% or 70% of data for the validation set - the validation set seems to play just as much role in the final estimate as the training data.
Early Stopping with Non-convex Objectives
The experiment above seems to demonstrate that early stopping should be almost certainly useless (if not harmful) for fitting simple convex models. However, it is never used with such models in practice. Instead, it is most often applied to the training of multilayer neural networks. Could it be the case that the method somehow becomes useful when the objective is highly non-convex? Let us run a small experiment, measuring the benefits of early stopping for fitting a convolutional neural-network on the MNIST dataset. For simplicity, I took the standard example from the Keras codebase, and modified it slightly. Here is the result we get when training the the most basic model:
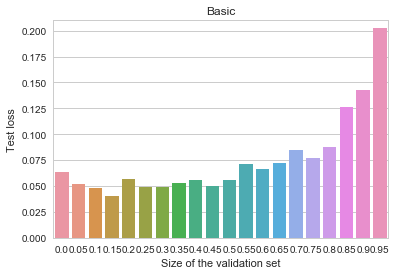
MNIST - Basic
The y axis depicts log-loss on the 10k MNIST test set, the x axis shows the proportion of the 60k MNIST training set set aside for early stopping. Ignoring small random measurement noise, we may observe that using early stopping with about 10% of the training data does seem to convey a benefit. Thus, contrary to our previous primitive example, when the objective is complex, early stopping does work as a regularization method. Why and how does it work here? Here's one intuition I find believable (there are alternative possible explanations and measurements, none of which I find too convincing or clear, though): stopping the training early prevents the algorithm from walking too far away from the initial parameter values. This limits the overall space of models and is vaguely analogous to suppressing the norm of the parameter vector. In other words, early stopping resembles an ad-hoc version of  regularization.
regularization.
Indeed, observe how the use of early stopping affects the results of fitting the same model with a small  -penalty added to the objective:
-penalty added to the objective:
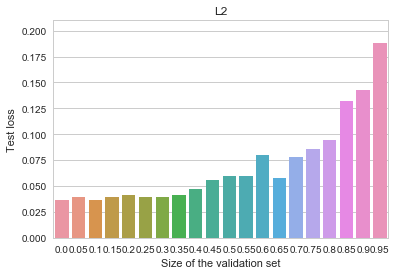
MNIST - L2
All of the benefits of early stopping are gone now, and the baseline (non-early-stopped, -regularized) model is actually better overall than it was before. Let us now try an even more heavily regularized model by adding dropout (instead of the penalty), as is customary for deep neural networks. We can observe an even cleaner result:
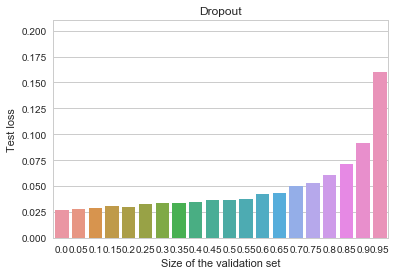
MNIST - Dropout
Early stopping is again not useful at all, and the overall model is better than all of our previous attempts.
Conclusion: Do We Need Early Stopping?
Given the reasoning and the anecdotal experimental evidence above, I personally tend to think that beliefs in the usefulness of early stopping (in the context of neural network training) may be well overrated. Even if it may improve generalization for some nonlinear models, you would most probably achieve the same effect more reliably using other regularization techniques, such as dropout or a simple penalty.
Note, though, that there is a difference between early stopping in the context of neural networks and, say, boosting models. In the latter case early stopping is actually more explicitly limiting the complexity of the final model and, I suspect, might have a much more meaningful effect. At least we can't directly carry over the experimental examples and results in this blog post to that case.
Also note, that no matter whether early stopping helps or harms the generalization of the trained model, it is still a useful heuristic as to when to stop a lengthy training process automatically if we simply need results that are good enough.
![\[\mathrm{cov}(X,Y) = E[(X - E[X])(Y - E[Y])].\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-1831261b0c8298b6a3d12ddea0042089_l3.png "Rendered by QuickLaTeX.com")
 , and stop there. Later on the covariance matrix would pop up here and there in seeminly random ways. In one place you would have to take its inverse, in another - compute the eigenvectors, or multiply a vector by it, or do something else for no apparent reason apart from "that's the solution we came up with by solving an optimization task".
, and stop there. Later on the covariance matrix would pop up here and there in seeminly random ways. In one place you would have to take its inverse, in another - compute the eigenvectors, or multiply a vector by it, or do something else for no apparent reason apart from "that's the solution we came up with by solving an optimization task". has a normal (or Gaussian) distribution with mean
has a normal (or Gaussian) distribution with mean  and covariance
and covariance  if:
if:![\[\Pr({\bf x}) =|2\pi{\bf\Sigma}|^{-1/2} \exp\left(-\frac{1}{2}({\bf x} - {\bf\mu})^T{\bf\Sigma}^{-1}({\bf x} - {\bf \mu})\right).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-c72dc0af31078a6b5c06ca187d4dd3e6_l3.png "Rendered by QuickLaTeX.com")
 ) and refrain from writing out the normalizing constant
) and refrain from writing out the normalizing constant  . Now, the definition of the (centered) multivariate Gaussian looks as follows:
. Now, the definition of the (centered) multivariate Gaussian looks as follows:![\[\Pr({\bf x}) \propto \exp\left(-0.5{\bf x}^T{\bf\Sigma}^{-1}{\bf x}\right).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-a5145d00c2add0f3651f234371825cbc_l3.png "Rendered by QuickLaTeX.com")
 (the identity matrix). Let us take a sample from it, which will of course be a symmetric, round cloud of points:
(the identity matrix). Let us take a sample from it, which will of course be a symmetric, round cloud of points: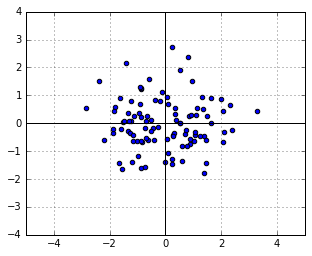
![\[P({\bf x}) \propto \exp(-0.5 {\bf x}^T {\bf x}).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-8a2ea394bef3046bc7e53d585738a238_l3.png "Rendered by QuickLaTeX.com")
 to the points, i.e. let
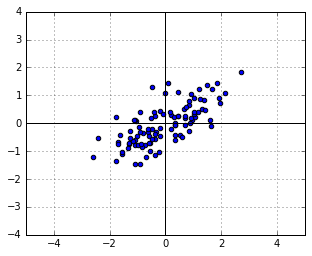
to the points, i.e. let  . Suppose that, for the sake of this example,
. Suppose that, for the sake of this example,  :
:
 into (1), to get:
into (1), to get:
 . The logic works both ways: if we have a Gaussian distribution with covariance
. The logic works both ways: if we have a Gaussian distribution with covariance  , and we are given
, and we are given  .
. , we can say that if our data were Gaussian, then it could have been obtained from a symmetric cloud using some transformation
, we can say that if our data were Gaussian, then it could have been obtained from a symmetric cloud using some transformation  , and we just estimated the matrix
, and we just estimated the matrix  for all rotation matrices. When we see a unit covariance matrix we really do not know, whether it is the “originally symmetric” distribution, or a “rotated symmetric distribution”. And we should not really care - those two are identical.
for all rotation matrices. When we see a unit covariance matrix we really do not know, whether it is the “originally symmetric” distribution, or a “rotated symmetric distribution”. And we should not really care - those two are identical.![\[{\bf \Sigma} = {\bf VDV}^T,\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-77a7920779e813aa9d2a69434934a91c_l3.png "Rendered by QuickLaTeX.com")
 is orthogonal (i.e. a rotation) and
is orthogonal (i.e. a rotation) and  is diagonal (i.e. a coordinate-wise scaling). If we rewrite it slightly, we will get:
is diagonal (i.e. a coordinate-wise scaling). If we rewrite it slightly, we will get:![\[{\bf \Sigma} = ({\bf VD}^{1/2})({\bf VD}^{1/2})^T = {\bf AA}^T,\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-2e20d88c4a3f98b68c62c23a2538d7a6_l3.png "Rendered by QuickLaTeX.com")
 . This, in simple words, means that any covariance matrix
. This, in simple words, means that any covariance matrix  followed by a rotation
followed by a rotation  . Just like in our example with
. Just like in our example with  above.
above. and thus contribute most to the spread of the data now. Can we only leave those and throw the rest out?
and thus contribute most to the spread of the data now. Can we only leave those and throw the rest out? as a
as a  and
and  , their
, their ![\[\langle {\bf v}, {\bf w}\rangle_{\Sigma^{-1}} = {\bf v}^T{\bf \Sigma}^{-1}{\bf w}.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-ccde20d05d63e8aeddefc2f4e97c1bab_l3.png "Rendered by QuickLaTeX.com")
![\[|{\bf v}|_{\Sigma^{-1}} = \sqrt{{\bf v}^T{\bf \Sigma}^{-1}{\bf v}},\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-f724b1da4af1628a260d50940cbee9d0_l3.png "Rendered by QuickLaTeX.com")
![\[|{\bf v}-{\bf w}|_{\Sigma^{-1}} = \sqrt{({\bf v}-{\bf w})^T{\bf \Sigma}^{-1}({\bf v}-{\bf w})}.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-99ad5ced1b604342ad5cd31d96b9c77a_l3.png "Rendered by QuickLaTeX.com")
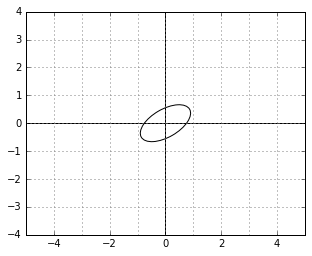 And here is an example of two vectors, which are considered “orthogonal”, a.k.a. “perpendicular” in this strange world:
And here is an example of two vectors, which are considered “orthogonal”, a.k.a. “perpendicular” in this strange world: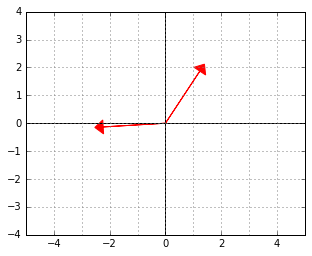
![\[{\bf v}^T{\bf \Sigma}^{-1}{\bf w} = {\bf v}^T({\bf AA}^T)^{-1}{\bf w}=({\bf A}^{-1}{\bf v})^T({\bf A}^{-1}{\bf w}),\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-de617b8c6664fb235e0fbc97b63fb9a3_l3.png "Rendered by QuickLaTeX.com")
 -“skewed” world. Somehow “deep down inside”, the ellipse thinks of itself as a circle and the two vectors behave as if they were (2,2) and (-2,2).
-“skewed” world. Somehow “deep down inside”, the ellipse thinks of itself as a circle and the two vectors behave as if they were (2,2) and (-2,2).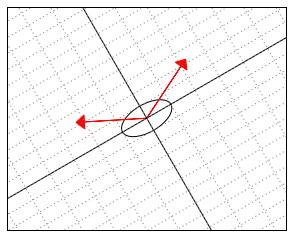
 . The PCA now becomes a method for analyzing the deformation of space, how cool is that.
. The PCA now becomes a method for analyzing the deformation of space, how cool is that. . Think of this inner product as a function
. Think of this inner product as a function  , which takes a vector
, which takes a vector  is known as the dual space to your “data space”.
is known as the dual space to your “data space”. an element of a dual space? Yes, it is, because, after all, it is a linear functional. However, the parameterization of this function is inconvenient, because, due to the skewed tensor, we cannot interpret it as projecting vectors upon
an element of a dual space? Yes, it is, because, after all, it is a linear functional. However, the parameterization of this function is inconvenient, because, due to the skewed tensor, we cannot interpret it as projecting vectors upon  . Instead, they satisfy
. Instead, they satisfy  . Things would therefore look much better if we parameterized our dual space differently. Namely, by considering linear functionals of the form
. Things would therefore look much better if we parameterized our dual space differently. Namely, by considering linear functionals of the form  . The new parameters
. The new parameters  could now indeed be interpreted as an “orthogonal direction” and things overall would make more sense.
could now indeed be interpreted as an “orthogonal direction” and things overall would make more sense.![\[f^{\Sigma^{-1}}_{\bf z}({\bf v}) = {\bf z}^T{\bf\Sigma}^{-1}{\bf v} = ({\bf \Sigma}^{-1}{\bf z})^T{\bf v} = f_{\bf w}({\bf v}),\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-84091d56adccf60763a2162ec33ba4bf_l3.png "Rendered by QuickLaTeX.com")
 . (Note that we can lose the transpose because
. (Note that we can lose the transpose because  . Or, in other words,
. Or, in other words, 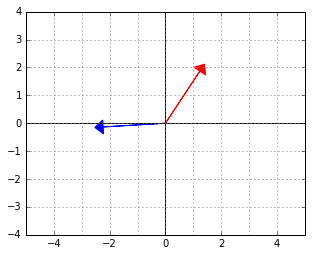
 . The red vector is an element of the “data space”, which would be mapped to 0 by this functional (because the two vectors are “orthogonal”, remember).
. The red vector is an element of the “data space”, which would be mapped to 0 by this functional (because the two vectors are “orthogonal”, remember).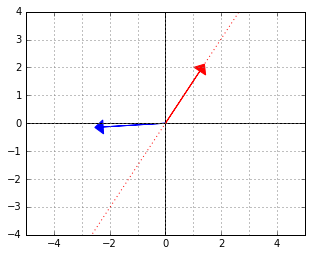
 will give us that normal. Here it is, the black arrow:
will give us that normal. Here it is, the black arrow: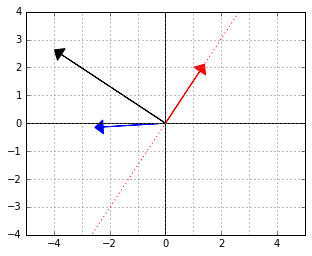
 or
or  , remember that those are simply inner products and (squared) distances in a skewed space, while
, remember that those are simply inner products and (squared) distances in a skewed space, while  are also quite common in practice. What about those?”
are also quite common in practice. What about those?” axis. The point (1,0) became (2,0), the point (3, 1) became (6, 1), etc. Think of it as changing the units of measurement - before we measured the
axis. The point (1,0) became (2,0), the point (3, 1) became (6, 1), etc. Think of it as changing the units of measurement - before we measured the  will be 1, because 2 pounds is still just 1 kilogram “deep down inside”.
will be 1, because 2 pounds is still just 1 kilogram “deep down inside”. , then
, then
 of the dual vectors. The metric tensor for the dual space must thus be:
of the dual vectors. The metric tensor for the dual space must thus be:![\[({\bf BB}^T)^{-1}=(({\bf A}^{-1})^T{\bf A}^{-1})^{-1}={\bf AA}^T={\bf \Sigma}.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-30e0bda6d93e7e5c5d06f3ee12f89c77_l3.png "Rendered by QuickLaTeX.com")
 metric? This is how the unit circle looks in the corresponding
metric? This is how the unit circle looks in the corresponding 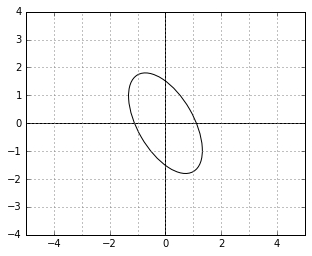
 is exactly the variance of the data along the direction
is exactly the variance of the data along the direction  ).
). (where
(where  is the data matrix) must be related to the
is the data matrix) must be related to the  .
. (vectors in the data matrix are row-wise, hence the multiplication on the right with a transpose). What is the covariance estimate of
(vectors in the data matrix are row-wise, hence the multiplication on the right with a transpose). What is the covariance estimate of  ?
?![\[{\bf Y}^T{\bf Y}/n=({\bf XA}^T)^T{\bf XA}^T/n={\bf A}({\bf X}^T{\bf X}){\bf A}^T/n\approx {\bf AA}^T,\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-7fe9f5f8b90f6c7b79a42c9aaf5aa003_l3.png "Rendered by QuickLaTeX.com")
 with uncorrelated variables of unit variance and then transformed using some matrix
with uncorrelated variables of unit variance and then transformed using some matrix  , and square it, the result can be regarded as a dot product of two "feature vectors", where the features are all pairwise products of the original inputs:
, and square it, the result can be regarded as a dot product of two "feature vectors", where the features are all pairwise products of the original inputs:
 to the third power, you are essentially computing a dot product within a space of all possible three-way products of your inputs, and so on, without ever actually having to see those features explicitly.
to the third power, you are essentially computing a dot product within a space of all possible three-way products of your inputs, and so on, without ever actually having to see those features explicitly. . We can "kernelize" it by first representing
. We can "kernelize" it by first representing  as a linear combination of the data points (this is called a
as a linear combination of the data points (this is called a ![\[f(x) = \left(\sum_i \alpha_i x_i\right)^T x + b = \sum_i \alpha_i (x_i^T x) + b,\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-21f9da75fd98376e9ab9bc50636bdd8e_l3.png "Rendered by QuickLaTeX.com")
 with a custom kernel function:
with a custom kernel function:![\[f(x) = \sum_i \alpha_i k(x_i,x) + b.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-2d96e2df41fbf5c0d519953b3871c6f1_l3.png "Rendered by QuickLaTeX.com")
 here, our model becomes a second degree polynomial regression. If
here, our model becomes a second degree polynomial regression. If  it is the fifth degree polynomial regression, etc. It's like magic, you plug in different functions and things just work.
it is the fifth degree polynomial regression, etc. It's like magic, you plug in different functions and things just work. , and, of course, the Gaussian function is one of these choices:
, and, of course, the Gaussian function is one of these choices:![\[k(x, y) = \exp\left(-\frac{|x - y|^2}{2\sigma^2}\right).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-fb0c759e10e4ae0cffed0a50acb032e1_l3.png "Rendered by QuickLaTeX.com")
 makes a kernel with a feature space, which includes all
makes a kernel with a feature space, which includes all  , for example. It is not hard to see that it corresponds to an inner product of feature vectors of the form
, for example. It is not hard to see that it corresponds to an inner product of feature vectors of the form![\[(x_1, x_2, \dots, x_n, \quad x_1x_1, x_1x_2,\dots,x_ix_j,\dots, x_n x_n),\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-99f7d81781e83520db1e527a74d645c4_l3.png "Rendered by QuickLaTeX.com")
 is also meaningful. It corresponds to scaling the corresponding features by
is also meaningful. It corresponds to scaling the corresponding features by  . For example,
. For example,  .
.![\[k(x,y) = 1 + x^Ty + \frac{(x^Ty)^2}{2} + \frac{(x^Ty)^3}{6}.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-66395891b5a5e441b149a82dc978ef67_l3.png "Rendered by QuickLaTeX.com")
 and all triple products scaled down by
and all triple products scaled down by  .
.![\[\sum_{i=0}^\infty \frac{(x^Ty)^i}{i!} = \exp(x^Ty).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-169bb5c3a324c272fb520911faef293c_l3.png "Rendered by QuickLaTeX.com")
 is a valid kernel function, which corresponds to a feature space, which includes products of input features of any degree, up to infinity.
is a valid kernel function, which corresponds to a feature space, which includes products of input features of any degree, up to infinity. before feeding it to the model. This is quite often a smart idea, which improves generalization. It turns out we can do this “data normalization” without really touching the data points themselves, but by only tuning the kernel instead.
before feeding it to the model. This is quite often a smart idea, which improves generalization. It turns out we can do this “data normalization” without really touching the data points themselves, but by only tuning the kernel instead. . If we normalize the vectors before taking their inner product, we get
. If we normalize the vectors before taking their inner product, we get![\[\left(\frac{x}{|x|}\right)^T\left(\frac{y}{|y|}\right) = \frac{x^Ty}{|x||y|} = \frac{k(x,y)}{\sqrt{k(x,x)k(y,y)}}.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-86156f93aa84c42485f78870af9a39f9_l3.png "Rendered by QuickLaTeX.com")

 in the denominator but by now you hopefully see that adding it is equivalent to scaling the inputs by
in the denominator but by now you hopefully see that adding it is equivalent to scaling the inputs by 
 before normalization). Simple, right?
before normalization). Simple, right? ,
,  . The value of the Gaussian kernel
. The value of the Gaussian kernel  for these inputs is:
for these inputs is:![\[k(x, y) = \exp(-0.5|1-2|^2) \approx 0.6065306597...\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-80d12497c000e4f901bd171a5db54e86_l3.png "Rendered by QuickLaTeX.com")
![\[\phi'(x) = \left(1, x, \frac{x^2}{\sqrt{2!}}, \frac{x^3}{\sqrt{3!}}, \frac{x^4}{\sqrt{4!}}, \frac{x^5}{\sqrt{5!}}\dots\right).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-3d661cde380f069bcf390789466e793e_l3.png "Rendered by QuickLaTeX.com")
 (1, 1, 0.707, 0.408, 0.204, 0.091, 0.037, 0.014, 0.005, 0.002, 0.001, 0.000, 0.000, ...)
(1, 1, 0.707, 0.408, 0.204, 0.091, 0.037, 0.014, 0.005, 0.002, 0.001, 0.000, 0.000, ...) (1, 2, 2.828, 3.266, 3.266, 2.921, 2.385, 1.803, 1.275, 0.850, 0.538, 0.324, 0.187, 0.104, 0.055, 0.029, 0.014, 0.007, 0.003, 0.002, 0.001, ...)
(1, 2, 2.828, 3.266, 3.266, 2.921, 2.385, 1.803, 1.275, 0.850, 0.538, 0.324, 0.187, 0.104, 0.055, 0.029, 0.014, 0.007, 0.003, 0.002, 0.001, ...) we just need to normalize:
we just need to normalize: (0.607, 0.607, 0.429, 0.248, 0.124, 0.055, 0.023, 0.009, 0.003, 0.001, 0.000, ...)
(0.607, 0.607, 0.429, 0.248, 0.124, 0.055, 0.023, 0.009, 0.003, 0.001, 0.000, ...) (0.135, 0.271, 0.383, 0.442, 0.442, 0.395, 0.323, 0.244, 0.173, 0.115, 0.073, 0.044, 0.025, 0.014, 0.008, 0.004, 0.002, 0.001, 0.000, ...)
(0.135, 0.271, 0.383, 0.442, 0.442, 0.395, 0.323, 0.244, 0.173, 0.115, 0.073, 0.044, 0.025, 0.014, 0.008, 0.004, 0.002, 0.001, 0.000, ...)![\[\scriptstyle\phi(1)^T\phi(2) = 0.607\cdot 0.135 + 0.607\cdot 0.271 + \dots = {\bf 0.6065306}602....\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-6c3c619ad7d23a5b02a9efb42ba5b9b9_l3.png "Rendered by QuickLaTeX.com")
 . The discrepancy is probably more due to lack of floating-point precision rather than to our approximation.
. The discrepancy is probably more due to lack of floating-point precision rather than to our approximation.
 ), hence these are not really all different features. Let us try to pack them more efficiently. As you'll see in a moment, this will open up a much nicer perspective on the feature vector in general.
), hence these are not really all different features. Let us try to pack them more efficiently. As you'll see in a moment, this will open up a much nicer perspective on the feature vector in general. must be repeated exactly
must be repeated exactly  times in our current feature vector. Thus, instead of repeating it, we could replace it with a single feature, scaled by
times in our current feature vector. Thus, instead of repeating it, we could replace it with a single feature, scaled by  . "Why the square root?" you might ask here. Because when combining a repeated feature we must preserve the overall vector norm. Consider a vector
. "Why the square root?" you might ask here. Because when combining a repeated feature we must preserve the overall vector norm. Consider a vector  , for example. Its norm is
, for example. Its norm is  , exactly the same as the norm of the single-element vector
, exactly the same as the norm of the single-element vector  .
.![\[\sqrt{\frac{n!}{a!b!}}\frac{x_1^a x_2^b}{\sqrt{n!}} = \frac{x_1^a x_2^b}{\sqrt{a!b!}} = \frac{x^a}{\sqrt{a!}}\frac{x^b}{\sqrt{b!}}.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-057fff7ee27f3b62cee6c7c547d67060_l3.png "Rendered by QuickLaTeX.com")
 = 231 features instead of 2097151. Nice!
= 231 features instead of 2097151. Nice!![\[\phi'_3(x_1, x_2) = \scriptstyle\left(1, x_1, x_2, \frac{x_1^2}{\sqrt{2!}}, \frac{x_1x_2}{\sqrt{1!1!}}, \frac{x^2}{\sqrt{2!}}, \frac{x_1^3}{\sqrt{3!}}, \frac{x_1^2x_2}{\sqrt{2!1!}}, \frac{x_1x_2^2}{\sqrt{1!2!}}, \frac{x_2^3}{\sqrt{3!}}\right).\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-c9783ab185dcb0ede49ad0adbaa145f4_l3.png "Rendered by QuickLaTeX.com")
 ,
,  (if we picked larger values, we would need to expand our feature vectors to a higher degree to get a reasonable approximation of the Gaussian kernel). Now:
(if we picked larger values, we would need to expand our feature vectors to a higher degree to get a reasonable approximation of the Gaussian kernel). Now: (1, 0.7, 0.2, 0.346, 0.140, 0.028, 0.140, 0.069, 0.020, 0.003),
(1, 0.7, 0.2, 0.346, 0.140, 0.028, 0.140, 0.069, 0.020, 0.003), (1, 0.1, 0.4, 0.007, 0.040, 0.113, 0.000, 0.003, 0.011, 0.026).
(1, 0.1, 0.4, 0.007, 0.040, 0.113, 0.000, 0.003, 0.011, 0.026). (0.768, 0.538, 0.154, 0.266, 0.108, 0.022, 0.108, 0.053, 0.015, 0.003),
(0.768, 0.538, 0.154, 0.266, 0.108, 0.022, 0.108, 0.053, 0.015, 0.003), (0.919, 0.092, 0.367, 0.006, 0.037, 0.104, 0.000, 0.003, 0.010, 0.024).
(0.919, 0.092, 0.367, 0.006, 0.037, 0.104, 0.000, 0.003, 0.010, 0.024). , what about the exact Gaussian kernel value?
, what about the exact Gaussian kernel value?![\[\exp(-0.5|x-y|^2) = 0.{\bf 81}87\dots.\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-937b3e338c9885a605aed33828832291_l3.png "Rendered by QuickLaTeX.com")
 -dimensional Gaussian kernel are:
-dimensional Gaussian kernel are:![\[\phi({\bf x})_{\bf a} = \prod_{i = 1}^d \frac{x_i^{a_i}}{\sqrt{a_i!}},\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-4b02f18f09aaa42f6c717d4606ec7fce_l3.png "Rendered by QuickLaTeX.com")
 .
. . Thus we may also state the following:
. Thus we may also state the following:![\[\phi({\bf x})_{\bf a} = \exp(-0.5|{\bf x}|^2)\prod_{i = 1}^d \frac{x_i^{a_i}}{\sqrt{a_i!}},\]](https://fouryears.eu/wp-content/ql-cache/quicklatex.com-206877da151c9fcd55ef7d50c5b279b3_l3.png "Rendered by QuickLaTeX.com")

 , where
, where  is the "fixed but unknown" parameter. Your whole "school of thought" is now focused on clever
is the "fixed but unknown" parameter. Your whole "school of thought" is now focused on clever  ,
,  , or
, or  . As a result, the probability distribution he works with are not parameterized any more, and all of the clever techniques that the classical statisticians have invented over the centuries for estimating parameters become seemingly useless. At this point a classical statistician puts his hands down and goes home, as there is nothing to do for him - there are no "unknowns". The Bayesian is, however, left to struggle with mathematically trivial, yet computationally incredibly heavy methods for extracting essentially the same values that the classical statistician could have obtained using his "parameter estimation" approaches. That's why the Bayesian "school of thought" is mostly focused on computationally-efficient methods for
. As a result, the probability distribution he works with are not parameterized any more, and all of the clever techniques that the classical statisticians have invented over the centuries for estimating parameters become seemingly useless. At this point a classical statistician puts his hands down and goes home, as there is nothing to do for him - there are no "unknowns". The Bayesian is, however, left to struggle with mathematically trivial, yet computationally incredibly heavy methods for extracting essentially the same values that the classical statistician could have obtained using his "parameter estimation" approaches. That's why the Bayesian "school of thought" is mostly focused on computationally-efficient methods for  and apply the Bayes rule here and there, whenever it seems appropriate. A number of computations derived from the two theoretical backgrounds end up exactly the same.
and apply the Bayes rule here and there, whenever it seems appropriate. A number of computations derived from the two theoretical backgrounds end up exactly the same.









